

Raw data from a sequencing facility are not trimmed. That means they contain the adapter sequences which were used to initiate the
DNA reproduction. RNAlysis can be used to generate trimmed from untrimmed data.
Command: FASTQ > Adapter trimming > Paired-end adapter trimming...
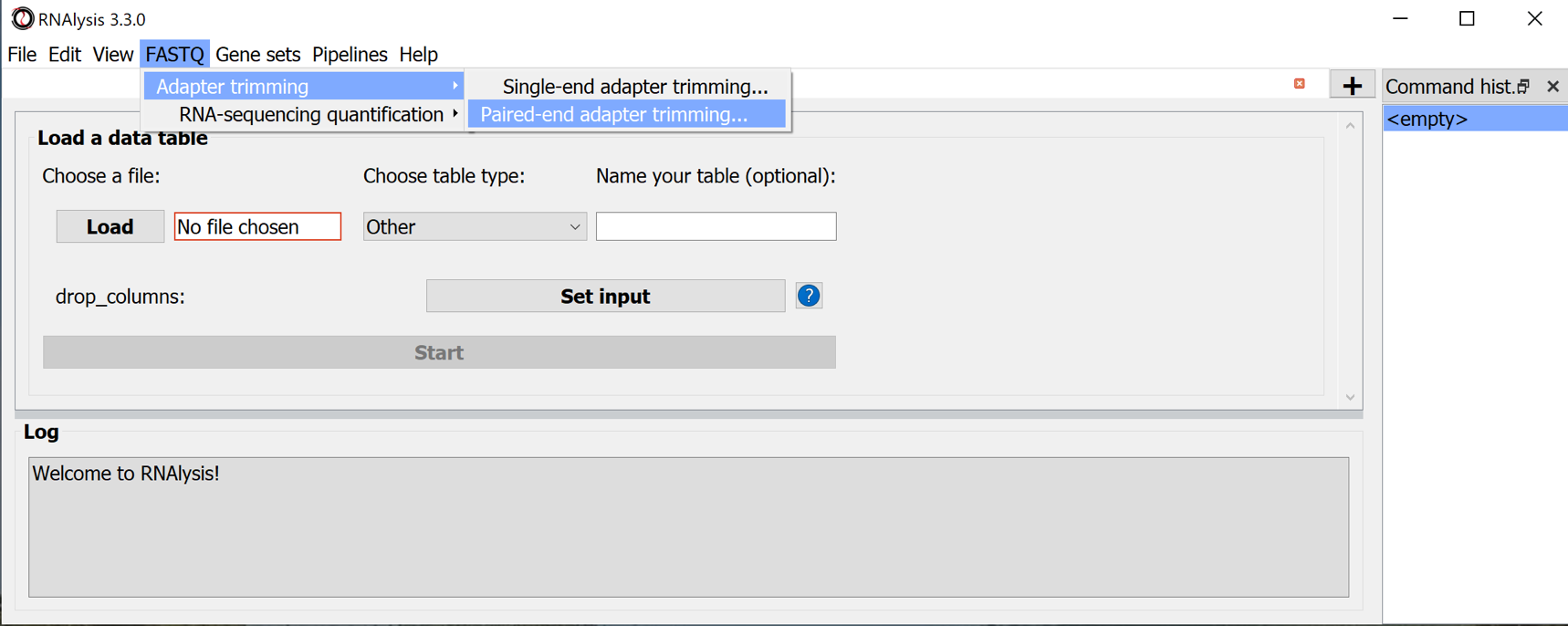
Parameters to set:
- output_folder
- R1 files and matching R2 files : the RNAseq data files
- Adapter sequences:
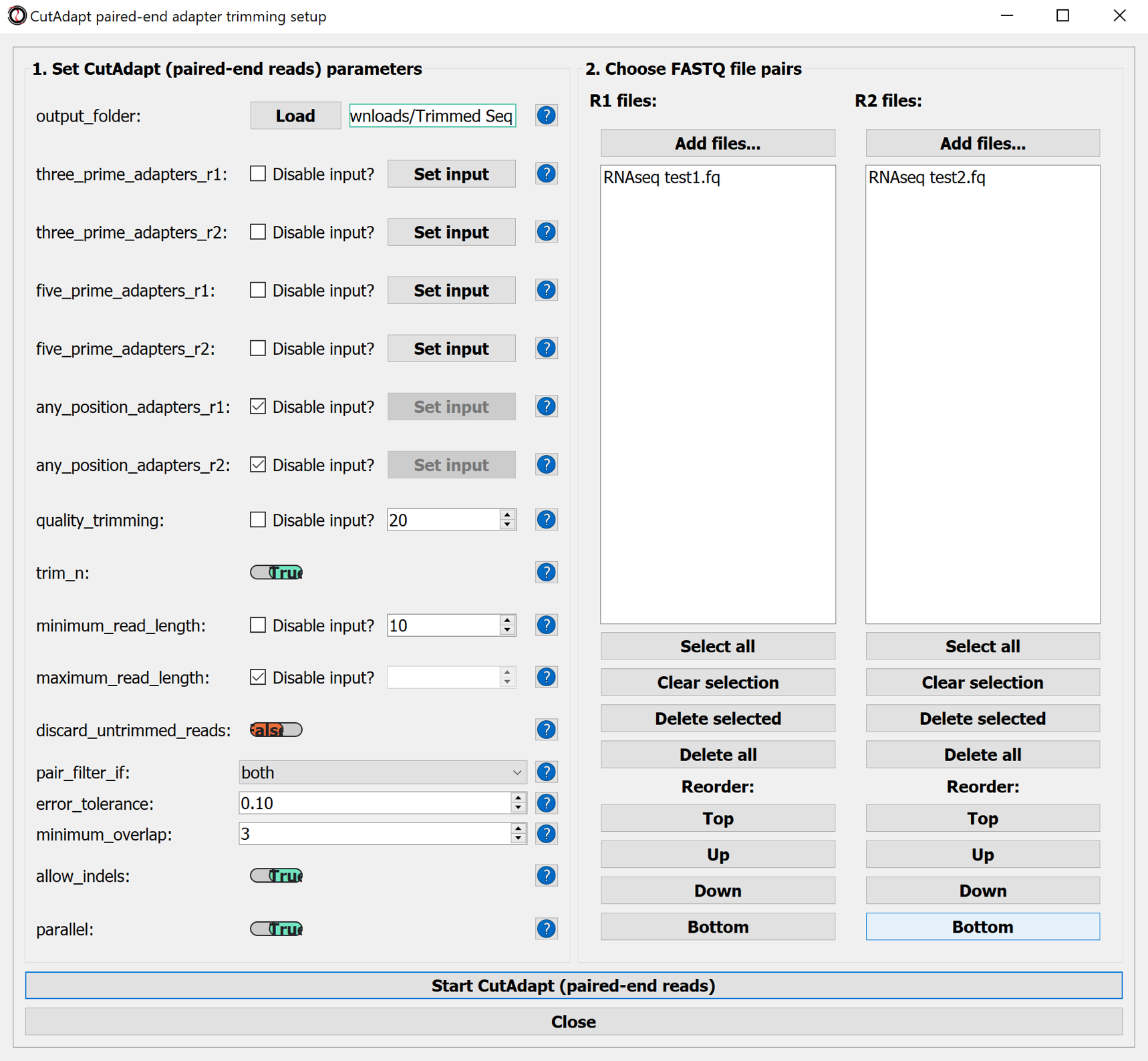
- three_prime_adapters_r1, - three_prime_adapters_r2, optionally: other adapter sequences
- Follow the original manual for the other parameters
- Use the button Start CutAdapt (paired-end reads) to start trimming
- Use the "Resource Monitor" application to monitor computer activity
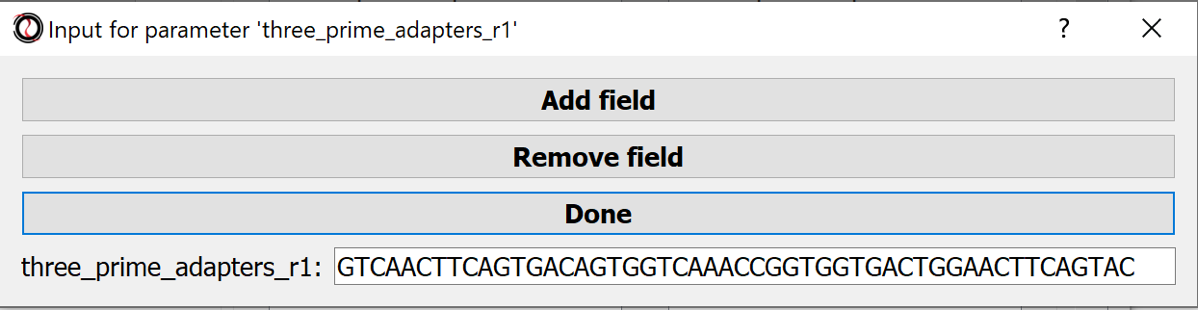
Results:
The result files should have roughly the same size as the untrimmed data files. Trimmed data files can be used to quantify the RNA expression levels.
Comments: matthias.wilm@ucd.ie
.